Article
Case Study: Capturing Quality Images for ML Algorithm

The FDA is currently collecting feedback on the draft guidance “Content of Premarket Submissions for Device Software Functions“, a highly anticipated draft to help update the guidance on software design file requirements based on risk.
Orthogonal is pleased to publish this guest blog by Brian Binkowski, who recently joined digital therapeutics (DTx) pioneer Happify (Congrats Brian!) after working at several MedTech companies, including Boston Scientific and Medtronic. Brian is an accomplished and impressive “friend of Orthogonal.” He shares our belief that the MedTech industry can rapidly improve patient outcomes by adopting fast feedback loop techniques to the development and enhancement of Software as a Medical Device (SaMD), DTx and connected medical device systems.

If you’re a technical person working at the intersection of regulatory, quality and software engineering in MedTech organizations and interested in the gritty details, then this article is a must-read. Have something to add? Feel free to reach out on our Contact Us page, or message Brian or Randy at their LinkedIn Pages.
To start this blog, I’ll share text from the FDA’s announcement of the document and highlight the most important information. Then, I’ll discuss what impact this document may have on those of us working with the development and manufacturing of SaMD.
The FDA’s Call for Comments on this draft guidance provides a concise overview of the contents of the actual document. Below, I pulled quotes from that announcement and added bold text for emphasis:

One of the key takeaways – and a major update from the previous guidance from 2005 – is the breakdown of the following Documentation Package Levels:
As with its predecessor, this updated guidance also provides the details of what should be included within each documentation level. However, I believe that one very useful and necessary thing that is missing from this new draft guidance is a cross-walk table that provides a correlation between the software safety classification and the directly related Documentation Levels.
To give an example of how the absence of this kind of cross-walk could lead to potential confusion, review the information in the “Software Testing as Part of Verification and Validation” row. (See Table 1.) In the new draft guidance, the Basic Documentation Level lists out unit and integration level testing descriptions to be included in the submission, but this is not consistent with the requirements in IEC 62304.[3] This could lead to situations where organizations – or multiple groups within one organization – follow the same guidance but end up with different sets of documentation for the same device, without a clear understanding of which set is appropriate.
(The good news in this particular example is that the bold “OR” statements in the “Software Development and Maintenance Practices” row allows conformance to IEC 62304. In other words, if you are already compliant with 62304, you don’t have to worry about a Basic vs. Enhanced level of documentation.)
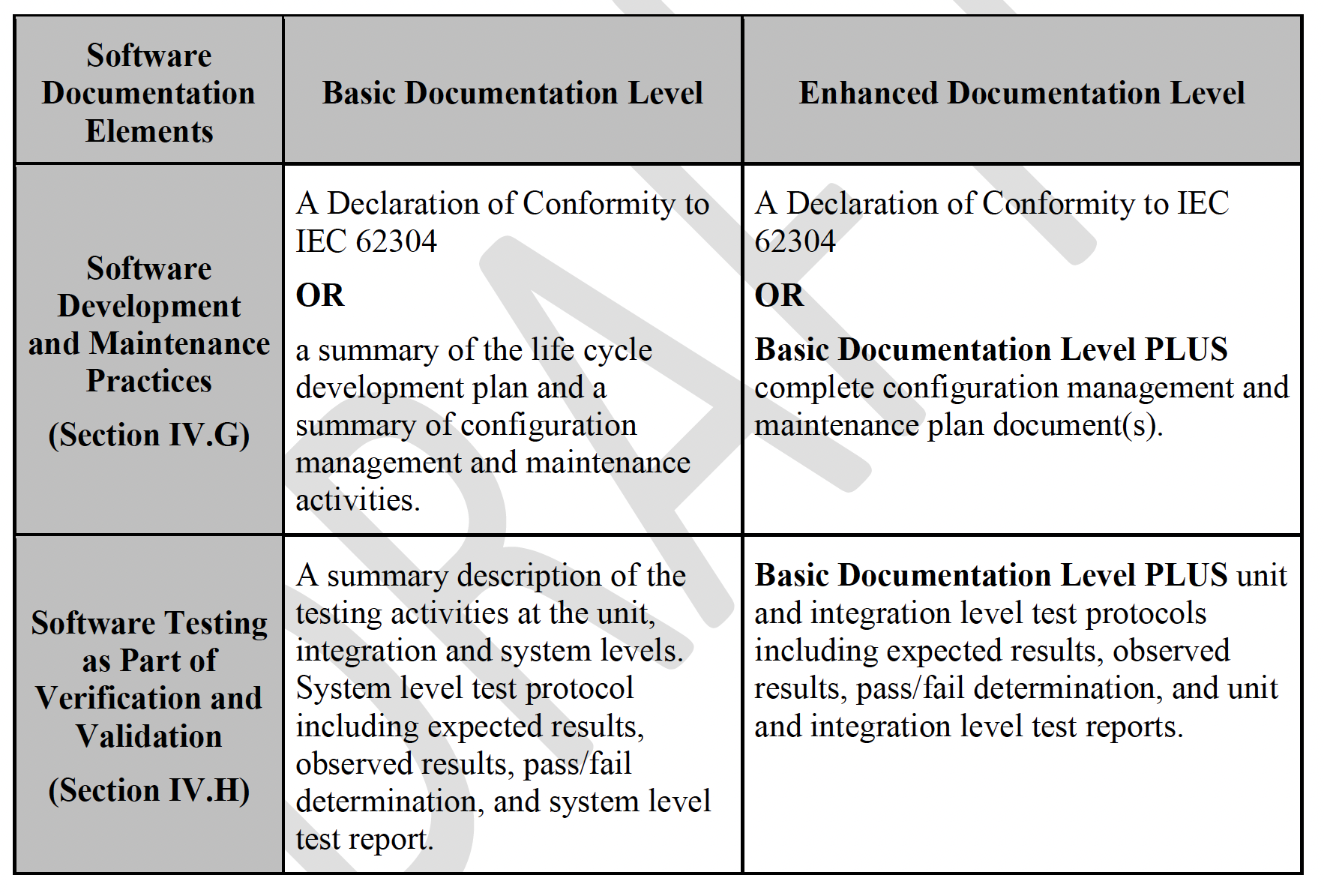
Table 1. Outline of Recommended Documentation, pages 9 and 10
Another area that caught my interest was the surprising callout to ANSI/AAMI SW91:2018, Classification Of Defects In Health Software.[4] To date, SW91 has not received much traction in the industry, and it is unclear how defect classifications are expected to be utilized as part of the submission process or post-market trending. It also came as a surprise that the reference to SW91 is within this guidance, but not referenced as part of the FDA Pre-Cert Pilot, which could be a significant step forward in helping to establish defect categories for the monitoring of industry-wide trends under Real World Performance Monitoring (RWPM).
By establishing clear defects that the industry can collectively trend, it sets the expectation that health software applications take defect trending seriously. For RWPM, the standardization of such defects could assist with clinical evaluations, change assessment and submission updates and future product submissions; existing data in the field can be utilized to demonstrate viable performance of the application.
Overall, I am excited to see how this document develops through the commenting period, and how it improves and gains more traction to help companies in this space.
Author’s Note: The opinions expressed in this blog are solely my own and do not express the views or opinions of Happify Health or any prior employers.
About Brian Binkowski
Brian Binkowski is a Director of Quality for Happify, a pioneering MedTech firm working in the Prescription Digital Therapeutic (PDT) space. Brian has deep experience working in the Digital Health space, and is passionate about Quality System Compliance in the new Digital era. He has previously worked at leading MedTech firms including Boston Scientific and Medtronic.
Happify’s Intelligent Healing Platform™ enables them to design personalized, science-based care solutions to improve health and well-being across the continuum of care—from digital therapeutics and coaching to community and wellness.
References: 1. This footnote is carried over from the FDA’s webpage: The term "device" is defined in 201(h) of the Federal Food, Drug, and Cosmetic (FD&C) Act to include an "instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is ...intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man ... or intended to affect the structure or any function of the body of man..." and "does not include software functions excluded pursuant to section 520(o) of the FD&C Act." 2. This footnote is carried over from the FDA’s webpage: See FDA website on "Software as a Medical Device (SaMD)." 3. IEC 62304:2006. ISO. https://www.iso.org/standard/38421.html. Published 2006. Accessed February 4, 2022. 4. ANSI/AAMI SW91:2018 - Classification of defects in health software. Webstore.ansi.org. https://webstore.ansi.org/standards/aami/ansiaamisw912018. Published 2018. Accessed February 4, 2022.
Related Posts
Article
Case Study: Capturing Quality Images for ML Algorithm

Article
Roundup: Bluetooth Medical Devices Cleared by FDA in 2024

Article
Help Us Build an Authoritative List of SaMD Cleared by the FDA
Article
SaMD Cleared by the FDA: The Ultimate Running List
